INTRODUCTION
Since 2015, the pharmaceutical industry has seen a significant increase in the design and trial of gene therapy drugs that are based on genetically modified microorganisms (GMM) (Figure 1 below). This is largely due to the clever application of genetic engineering which has created new biological tools such as Crispr/Casp 9 and replication incompetent target specific viral vectors. These tools are special as their application allows targeted delivery of gene therapy, therefore reducing the risk to the health of patients. This article talks about the challenges that pharmaceutical companies face in recruiting trial participants for the increasing number of GMM-based gene therapy clinical trials. Currently recruitment numbers are low and a key reason is the lack in capability of hospital and pharmaceutical settings to satisfy biosafety regulatory requirements. To address this, the article proposes a three-step plan to satisfy biosafety requirements. The plan is based on increasing biosafety awareness, training, and support to fulfil biosafety regulatory requirements. It has been documented that lacking the capabilities to satisfy biosafety regulatory requirements prevents hospital and pharmaciesfrom participating in clinical trial recruitment programmes. Therefore, if regulatory affairs experts(and others responsible for achieving buy-in of potential trial locations) work together with their stakeholders to fulfil the biosafety requirements, the challenge can be tackled to drive up numbers. This joint effort will in turn lead to more robust efficacy and patient safety data.
THE CHALLENGE
The best place to recruit patients is in hospital and convalescent settings. Increasing recruitment is particularly important when developing treatments for rare hereditary conditions. One example is spinal muscular atrophy (SMA) disease – which causes reduction in muscle function, impairs everyday vital activities such as eating and breathing, and quality of life is grossly impacted. Zolgensma® (Onasemnogene abeparvovec) is a gene therapy drug based on a genetically modified microorganism called adeno associated virus and has been approved for the treatment of SMA (Keinath). The adeno associated virus can deliver genes to target areas. In the case of Zolgensma, the adeno associated viral vector (AAV) delivers a working copy of the SMA gene to the cells that need it, therefore improving muscle function. This is a wonderful example of the successful application of gene therapy to treat disease, which would not have been possible without successful patient recruitment at the clinical trial stage. There are many other safe and effective gene therapy drugs being developed based on the AAV technology and other viral vectors. Each year, an increasing number of gene therapies enter the clinical trial stage (Figure 1).
The challenge for many is overcoming low patient recruitment numbers for upcoming gene therapy products that have the same potential for marketsuccess. We are in an age where gene therapy has become safer. Nevertheless, clinical trial sample sizes remain small. Almost half of gene therapy clinical trials had a sample size of less than 25 patients (Overbeke). A review highlighted that this problem is highest in Europe followed by the US and Canada (Overbeke). A barrier is lack of information about biosafety measures and legal notifications required for hospital and pharmacy settings to engage
in gene therapy trial studies. A survey of 1440 pharmacists in the United Kingdom (UK) revealed that lack of familiarity with gene therapies was reflected by trial low response rate (Petrich). In a study conducted by the American Society of Health-System Pharmacists, of the 3453 pharmacists surveyed, 37% of their study respondents were still planning where their gene therapy products would be stored, and 30% were unsure of whether they had the correct biosafety equipment (Petrich). Petrich et al. argue that “Ultimately, as more gene therapy products are approved, institutions that want to provide these advanced therapies to their patients must be prepared to develop their own policies until such guidelines are published.” Until these issues are resolved the challenge will persist.
In a UK scenario, a Contract Research Organisation was unsuccessful in recruiting pharmacies to participate in a clinical trial commission by a US pharmaceutical company. Communications between the contracted biosafety consultant and the hospital pharmacies showed that the pharmacy team expected a short site survey followed by a supply of drug products to begin the clinical trial. It is very likely that this situation is similar in settings across the UK and overseas, and highlights the need for biosafety awareness as it relates to clinical trials. In this scenario Petrich et al.’s ecommendation has some merit. They recommend “that clinical pharmacists lead in the formation of clinical biosafety committees if their institutions are expecting to administer any approved gene therapy products”. The foreseeable issue, however, is the demand on the clinical team’s time. This approach will require significant training
for the clinical pharmacist (and others involved in the process) to be experts on biosafety requirements.
Alternatively, the quicker and more thorough approach is to provide biosafety support through a regulatory affairs team, with the mission to make the process near effortless for clinicians and other hospital and pharmacy stakeholders. So what practical steps can be taken to facilitate satisfying biosafety requirements in order to increase patient recruitment into gene therapy drug trials?
THE THREE STEP PLAN
This three-step plan is specific to trials occurring in the UK. Other countries will have different legal requirements. In the UK, a few legislations apply to the use of gene therapy products that are based on the use of GMM. One key regulation is “The Genetically Modified Organisms (Contained Use) Regulations 2014” which is enforced by a regulatory body known as the Health and Safety Executive (HSE). This regulation requires the trial/study site to have the necessary containment level (biosafety level) measures of which there are 20 requirements to consider, as well as other controls including risk assessments.
Firstly, the site will need to notify the HSE’s bioagents division that its premises will be used for contained use work. The form must be accompanied by a risk assessment which has been reviewed by a competent biological safety officer/advisor, as well as by a Genetic Modification Safety Committee, depending on the level of risk. Since some shedding may occur once the drug is administered to the patient, The Genetically Modified Organisms (Deliberate Release) Regulations 2002 might need to be considered. This regulation is enforced by the Department for Environment, Food and Rural Affairs (Defra). The UK’s Scientific Advisory Committee on Genetic Modification (SACGM) has written “The SACGM Compendium of Guidance” to help stakeholders fulfil the requirements regarding clinical trial studies. The process of fulfilling all biosafety legislations requires a great deal of knowledge, expertise and most importantly, time. For this reason, the best persons to help study sites and pharmaceutical companies achieve biosafety compliance are experts in the field. Here is a three-step plan that pharmaceutical companies can take to achieve this.
1) Set the expectations with the hospital’s clinicians, pharmacists and involve their hospital’s health and safety team. Be open and clear during initial discussions. Be specific about the process that lies ahead. Clinicians and pharmacists care about patients and they want to go the extra mile to help them. Telling them in advance means that there are no surprises, and they will be more likely to commit. In this sector, surprises are undesirable and have led to nonstarter trials. Avoid this by providing quality upfront information. This approach will go a long way to convince your prospective clinical trial sites to take part in your study and to have confidence in you. It is a good idea to make this available in an easy-to-read pamphlet or guide which they can take away and process.
2) Connect your clinical trial site with a competent biosafety officer or consultant Now that you have set their expectations, clinicians will need to be assured that an expert is at hand to help them seamlessly navigate the process. Remember, your stakeholders are busy delivering on their day-to-day objectives,
including the completion of mountains of paperwork! Getting on board with your trial will impact on their limited resource. Your best chance of getting buy-in is to help them to visualise themselves being in the backseat of your Bentley (other cars are available). You do this by connecting them with an expert biosafety consultant who will be in the driver’s seat, taking the stress out of the process, by managing the stages, paperwork, and critical liaisons.
3) Flatten the speed Bumps
Now that they are on board, empower the biosafety consultant to chart the way. Too often, success is hindered by having to rely on third-party consent for every action. In clear terms, having every biosafety related piece of work go through a contract research organisation or the pharmaceutical company for approval, slows the pace of progress, makes for cumbersome dealings, and the decision to stay on board can change because of this. To avoid this, arrange a scope for work with your biosafety consultant, which is reasonably unrestrictive, whilst producing the desired effect. The regulations that the biosafety consultant will need to consider may include others in addition to those mentioned previously, so it is important to make the scope of work flexible enough for them to consider other regulatory requirements.
HORIZON SCANNING
The Development of gene therapy products based on viral vectors (derived from viruses) has been steadily increasing since 2003. Figure 1 shows the number of approved gene therapy products from 2003 – 2017 (Goswani). The timeline shows that since 2015 greater numbers of gene therapy products have been approved.

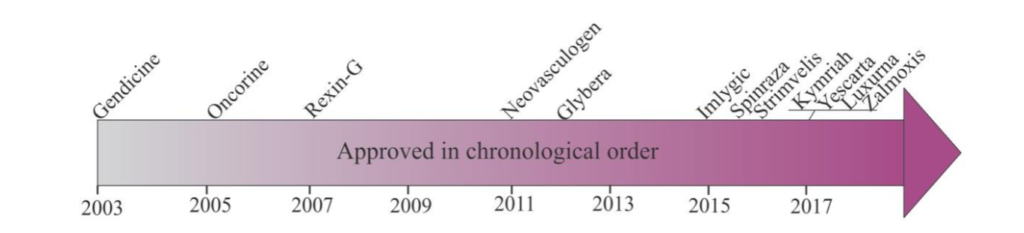
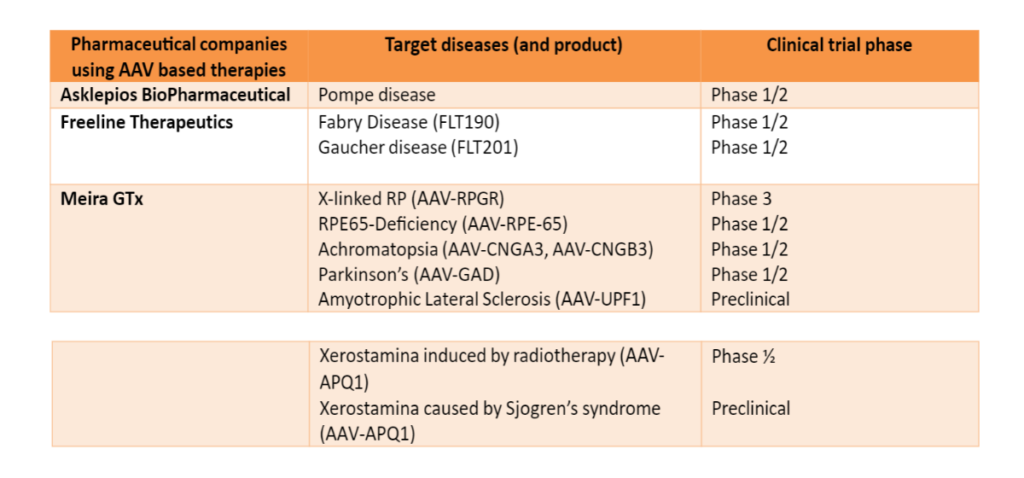
Table 1. Pharmaceutical companies with gene therapy products in clinical trial stages (data correct at 17th January 2023).
CONCLUSION
Gene therapy products for treatment of various diseases are continuously being developed. For clinical trials of these drugs to take place in pharmacy and hospital settings, these facilities will need to have the correct biosafety practices and facility requirements as specified by regulators of the country. There is evidence that patient enrolment numbers into clinical trials are low, due to clinicians and pharmacists being unaware of biosafety requirement. It is therefore vital that these stakeholders receive the knowledge and support needed to fulfil biosafety requirements so that enrolment numbers can be boosted. This will enable gathering of more robust data to sufficiently determine drug efficacy and safety.
References
Keinath M, Prior D, and Prior T. Spinal Muscular Atrophy: Mutations, Testing and Clinical Relevance. The Clinical Application of Genetics. 2021:14 11–25
Petrich J, Marchese D, Jenkins C, Storey M, Blind J. Gene Replacement Therapy: A primer for the health-system
Pharmacist. Journal of Pharmacy Practice. 2020;30(6):846-855.
Overbeke E, Michelsen S, Toumi, Steven H, Trusheim M, Huys I, Simoens. Market access of gene therapies across Europe USS and Canada: Challenges, trends and solutions. Drug discovery today. 2021;100(00):
Voysey M, et al. Single Dose Administration, And The Influence Of The Timing Of The Booster Dose On
Immunogenicity and Efficacy Of ChAdOx1 nCoV-19 (AZD1222) Vaccine. Reprints with the Lancet. 2021
Goswani R, Subramanian G, Silayeva L, Newlirk I, Doctor D, Chawla K, Chattopadhyay, Chandra D, Chilukuri N, Betapudi V. Gene therapy Leaves a Vicious Cycle. Frontiers in Oncology.2019;9(297):
FDA, https://www.fda.gov/news-events/press-announcements/statement-fda-commissioner-scott-gottlieb-md-andpeter-marks-md-phd-director-center-biologics, 17January 2023
MHRA, https://www.gov.uk/government/case-studies/horizon-scanning-case-study-developing-standards-foradeno-associated-virus-gene-therapies, 17 January 2023
With thanks to Bridget Leathley, CFIOSH and Audrey Williams for editorial input
Email: info@bishopsimon.co.uk
Note: All legislations and guidance are current at the time of writing (09 May 2022). Please ensure that you always consult the most recent legislation and guidance. © Dr Shurene Bishop Simon. May 2022. All rights reserved.